
Spatial Proteomic Analysis of Isogenic Metastatic Colorectal Cancer Cells Reveals Key Dysregulated Proteins Associated with Lymph Node, Liver, and Lung Metastasis

 , ,
, ,  , , , , ,
, , , , ,  and
and 
Abstract
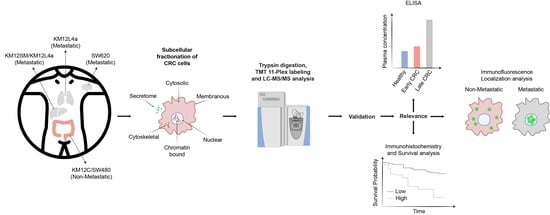
:
1. Introduction
2. Materials and Methods
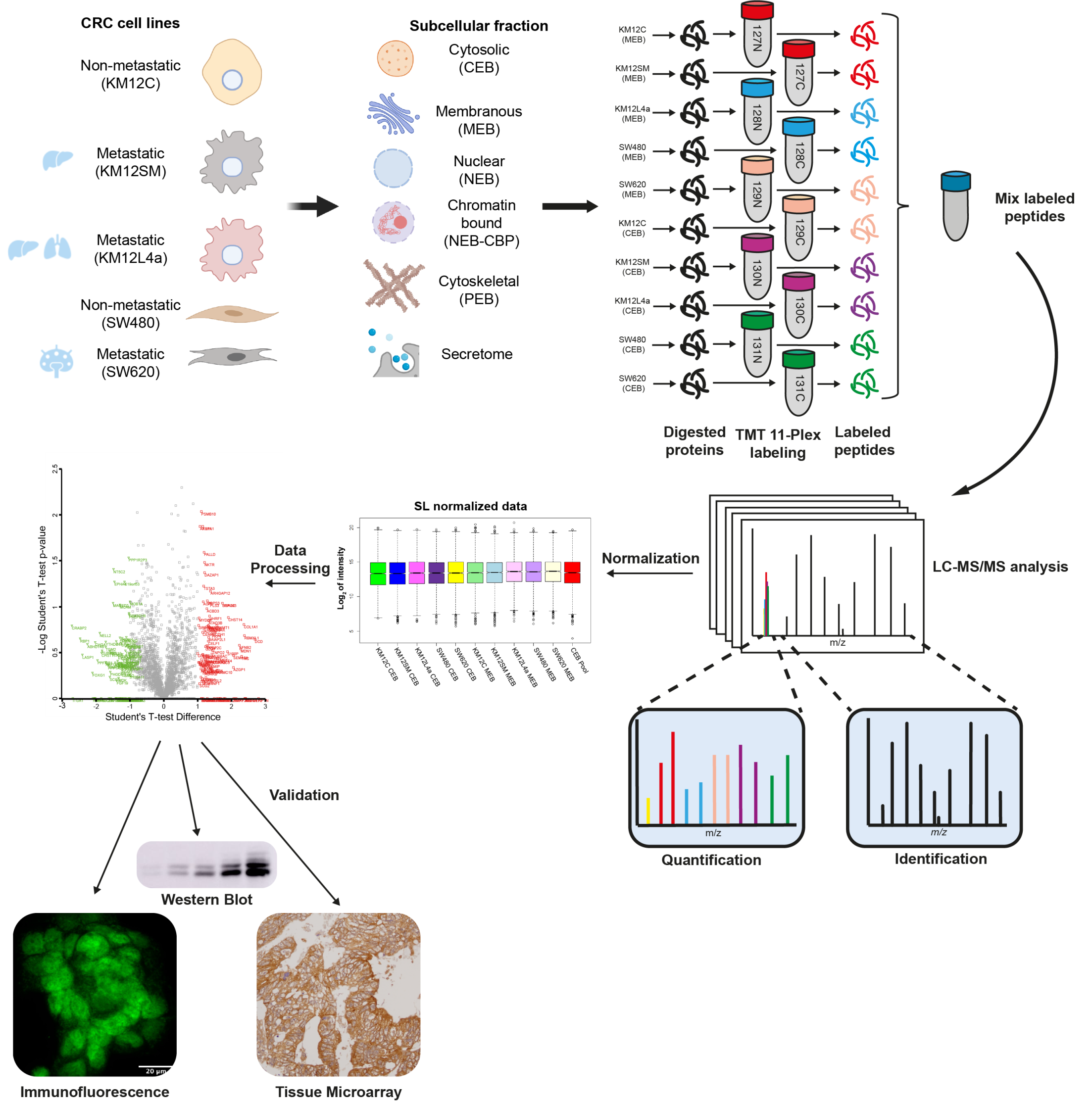
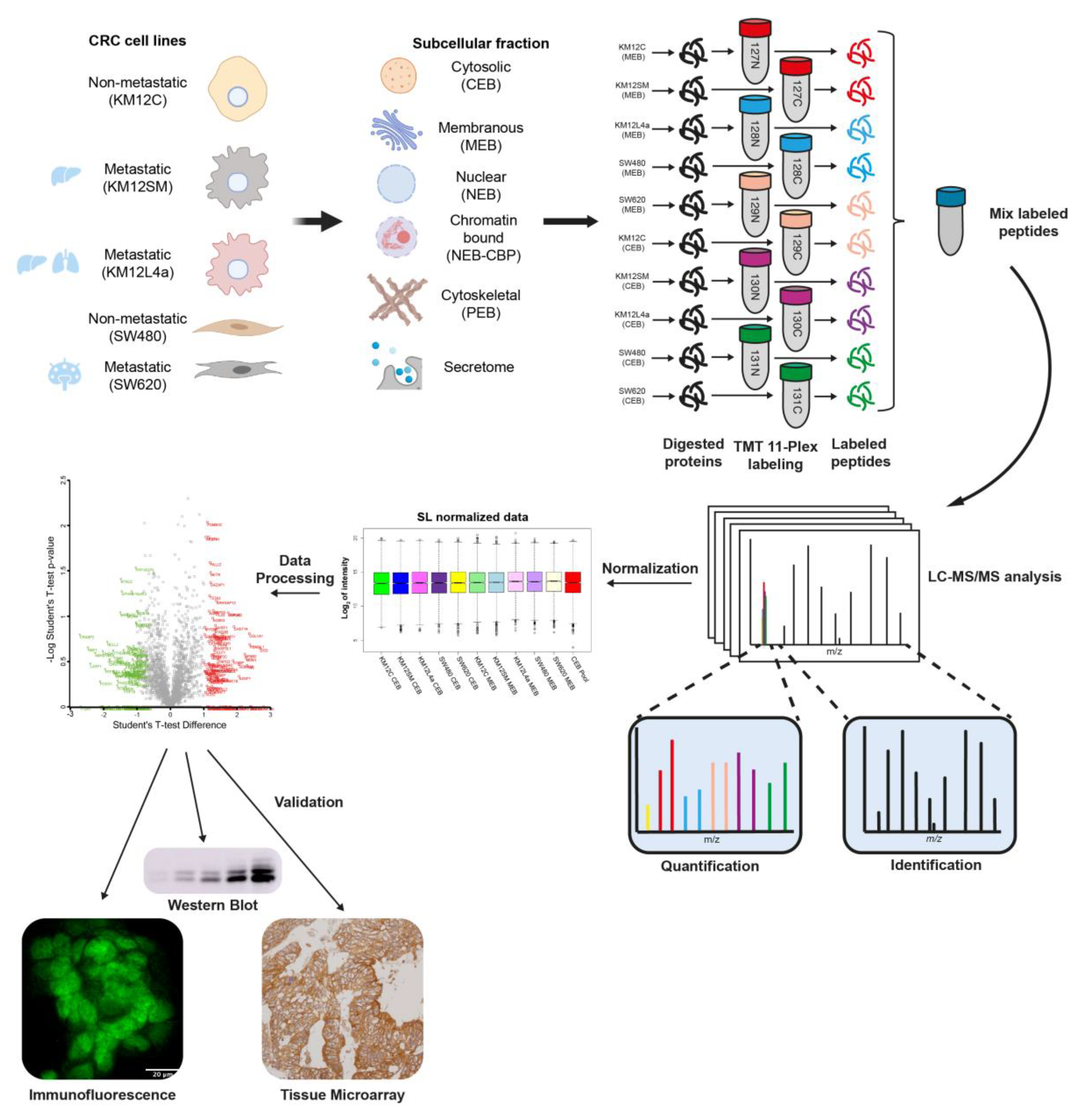
2.1. CRC Cell Lines
2.2. Protein Extracts and Quantification
2.3. TMT 11-Plex Labeling
2.4. LC–MS/MS Analysis
2.5. MS Data Analysis
2.6. Tissue and Plasma Samples
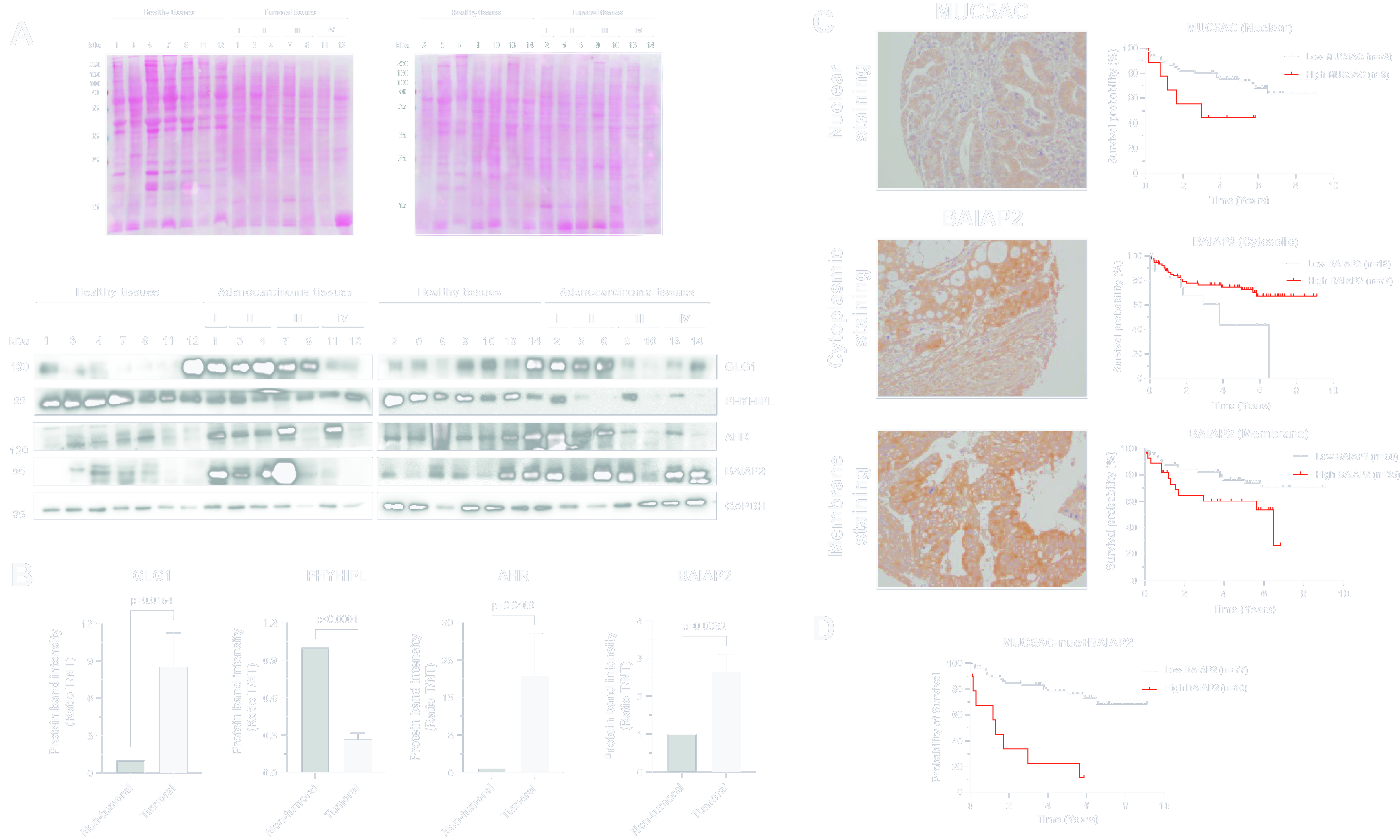
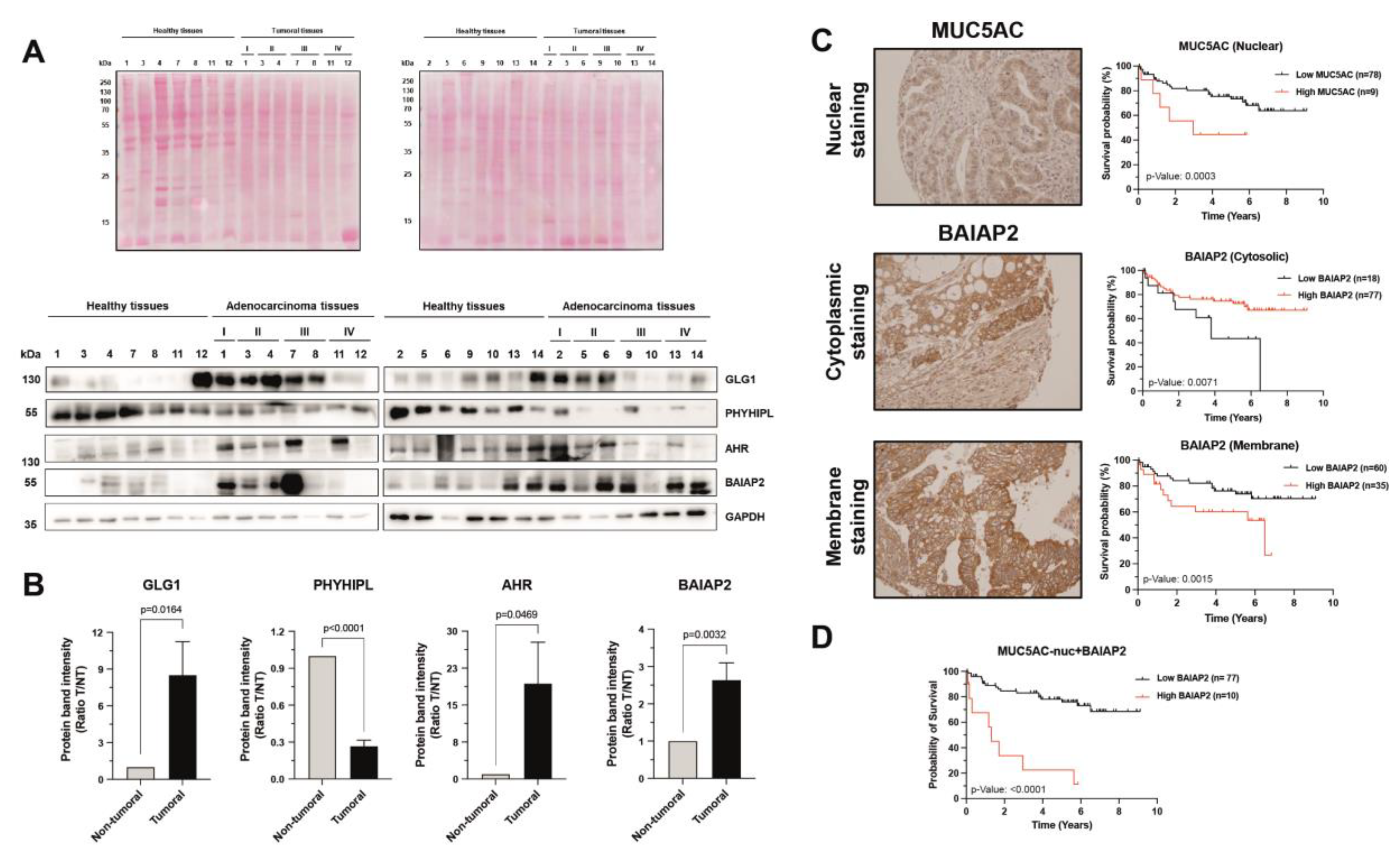
2.7. Western Blot
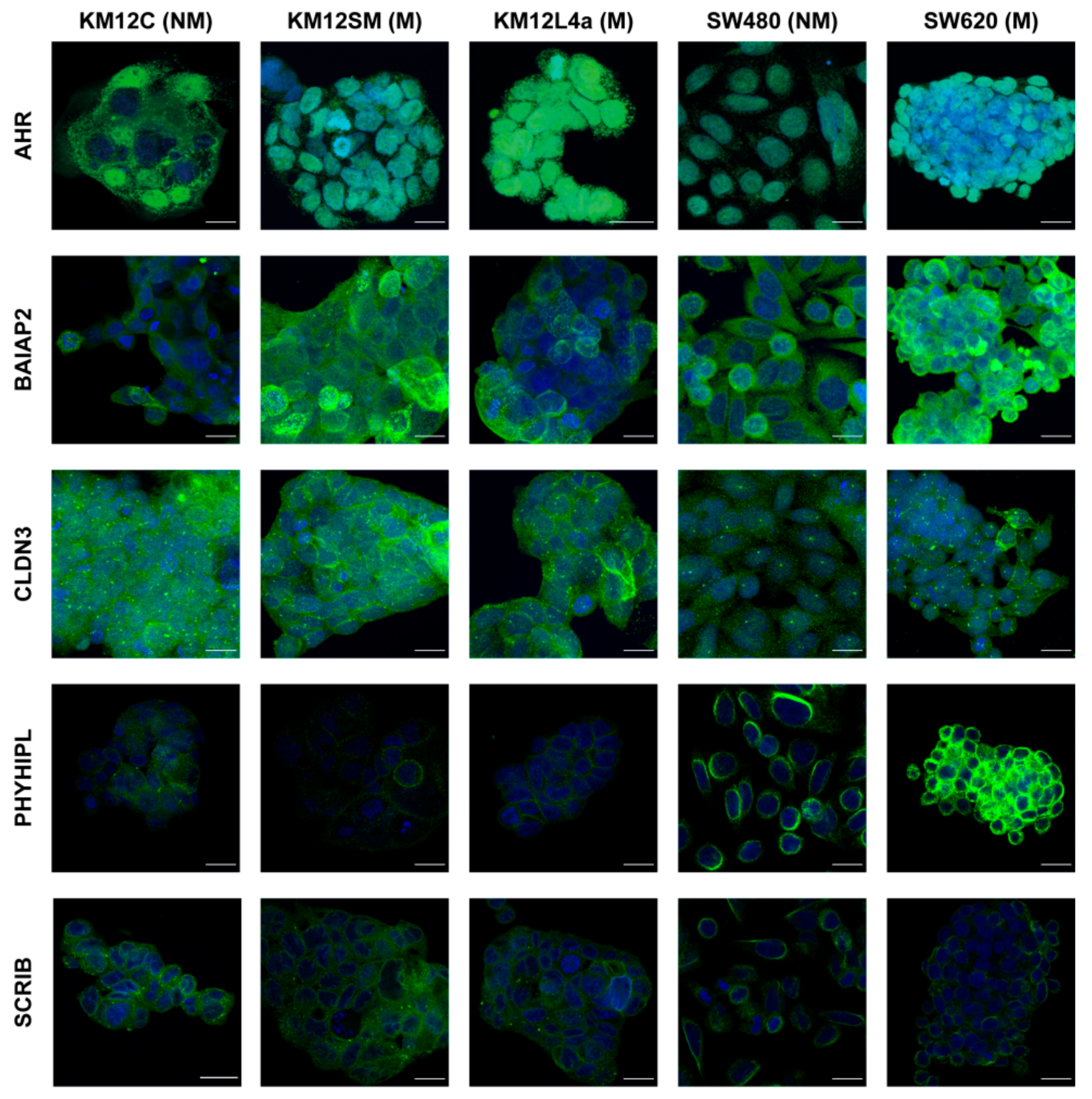
2.8. Immunofluorescence
2.9. In Silico Analysis
2.10. Immunohistochemistry, Tissue Microarrays and ELISA
2.11. Statistical Analysis
3. Results
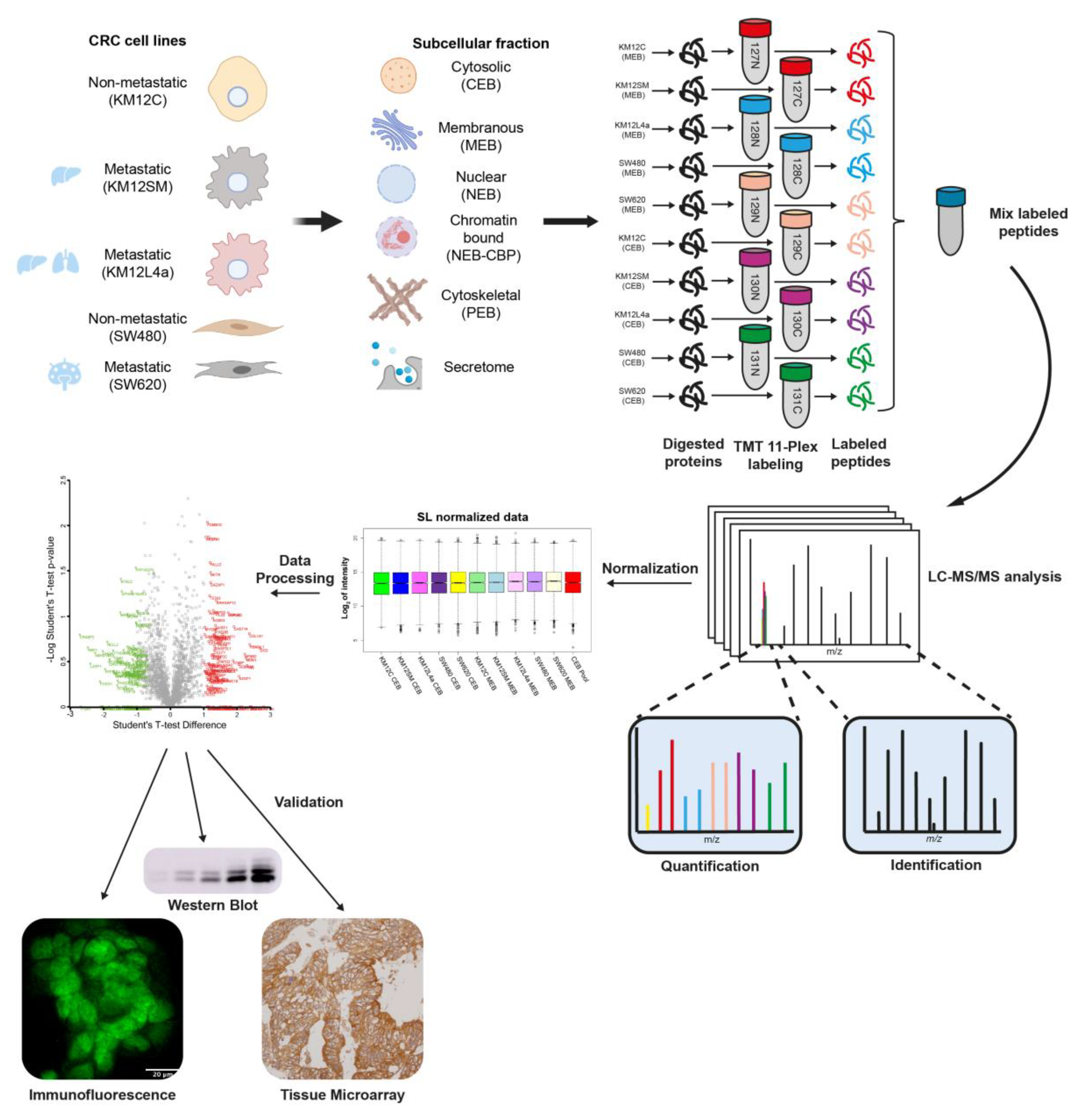
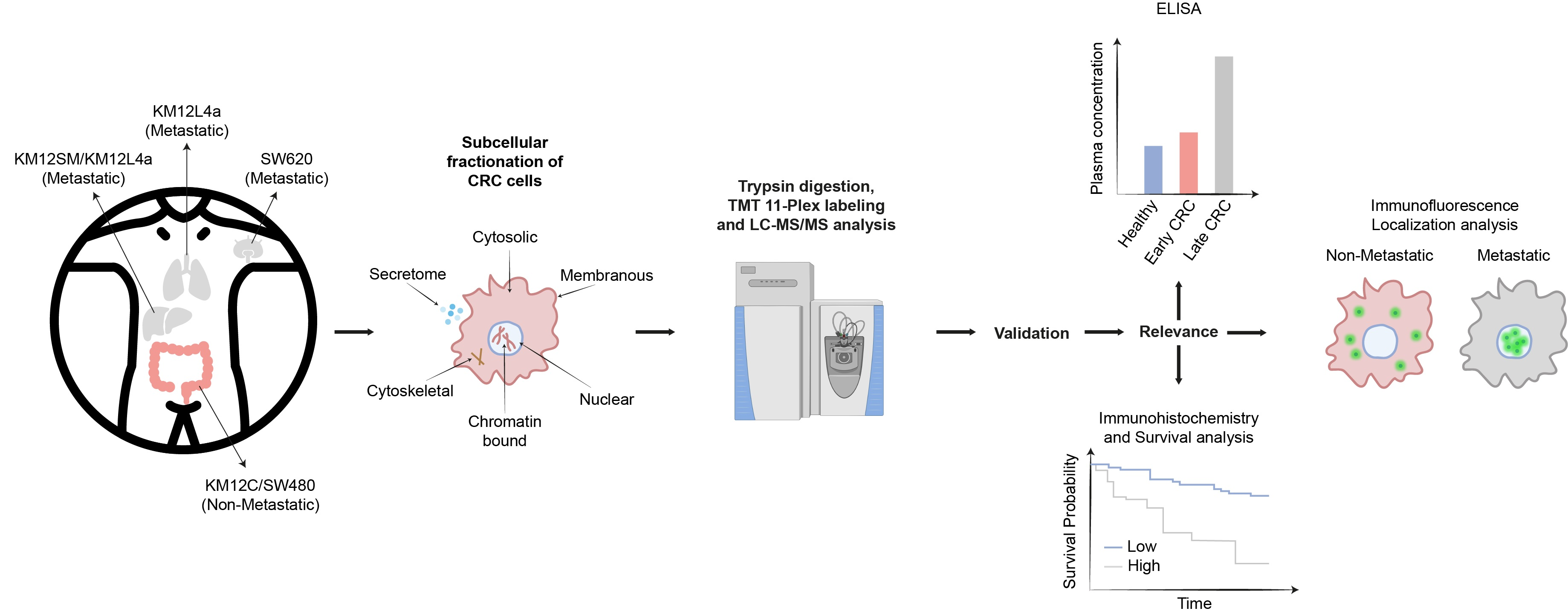
3.1. Subcellular Fractionation and Analysis for Differential Protein Expression and Localization in Lymph Nodes, Liver and Lung Metastatic Tropism of Colorectal Cancer Cells
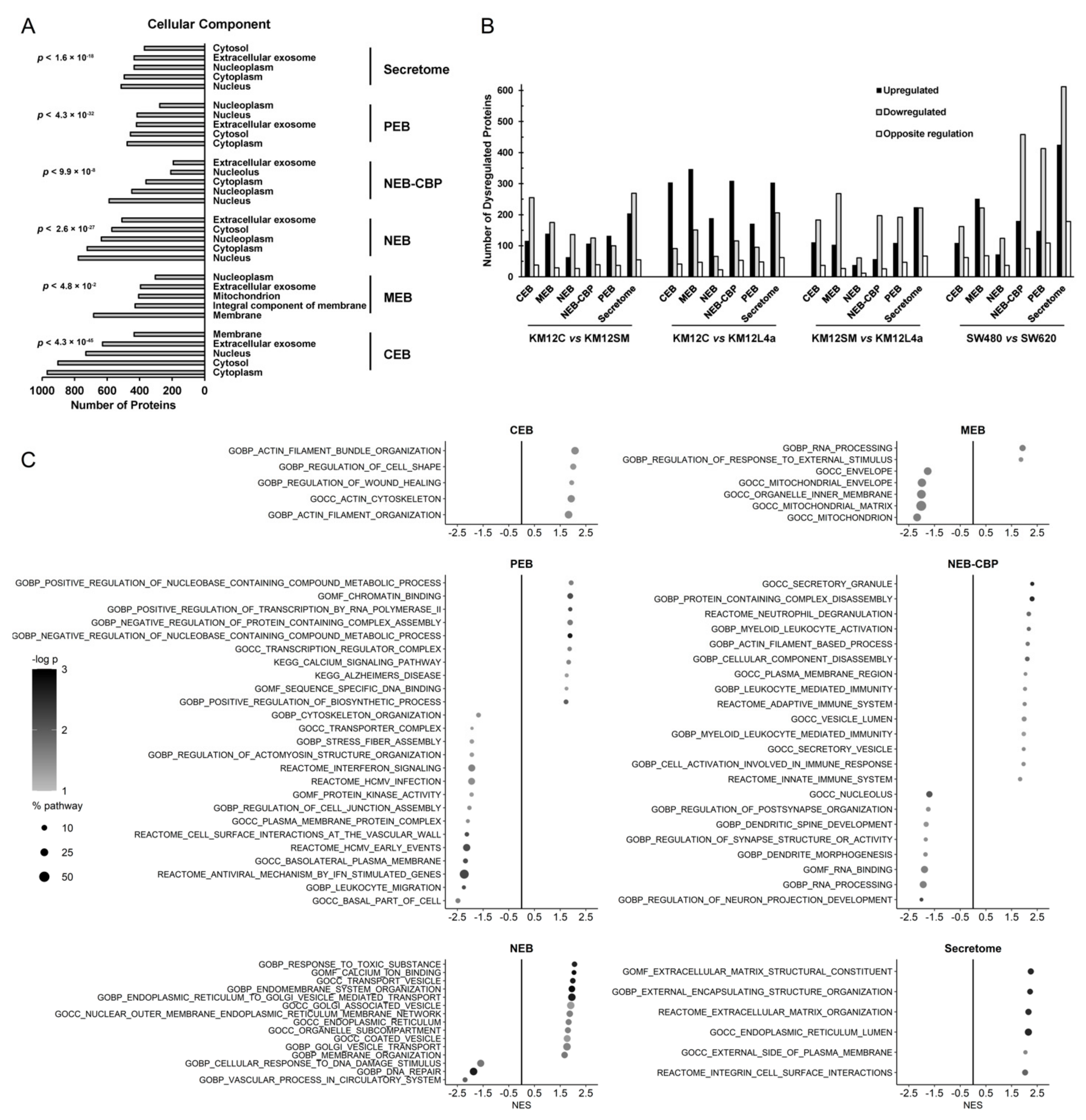
3.2. Mapping Spatial Protein Alterations in CRC Metastatic Cells
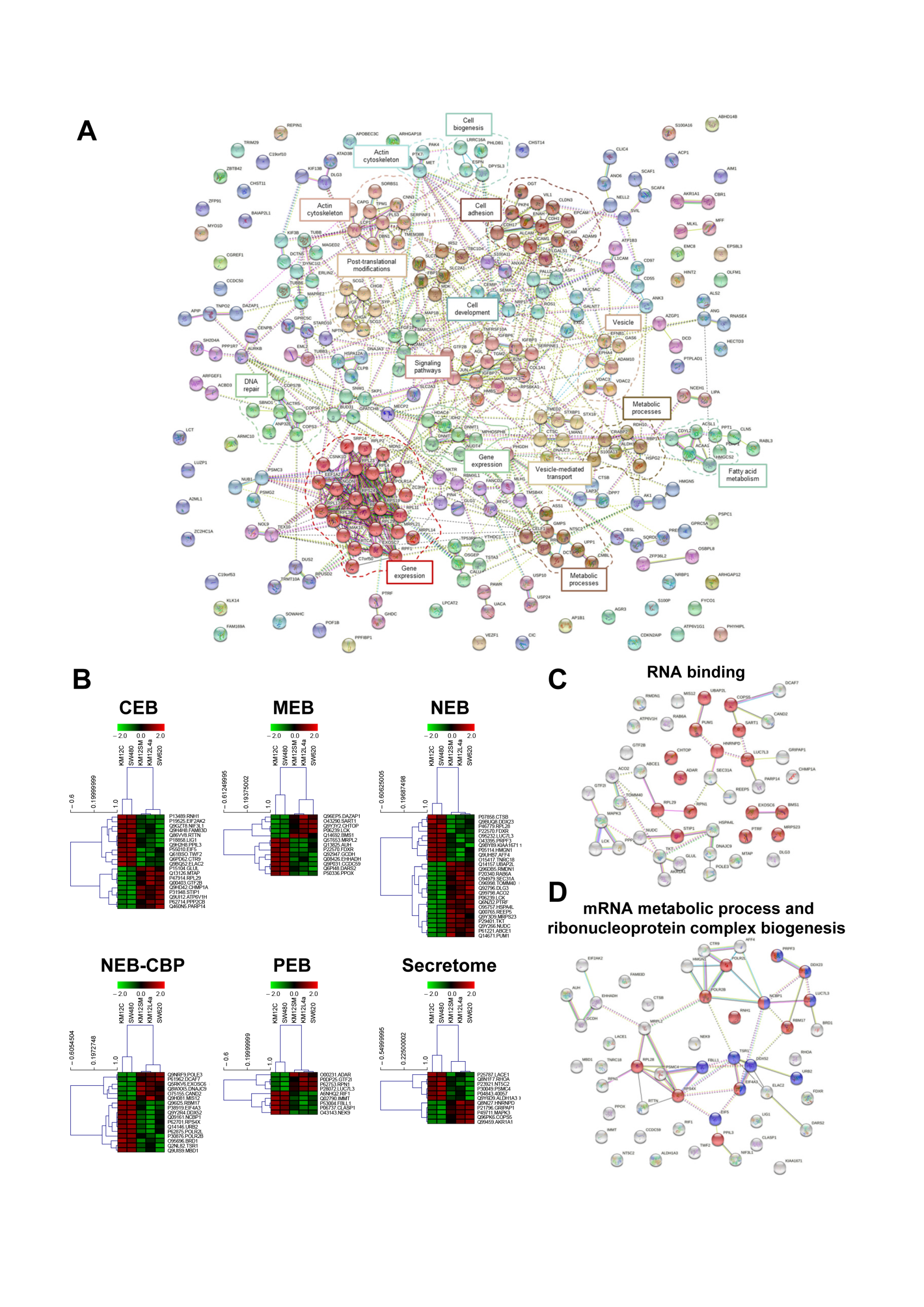
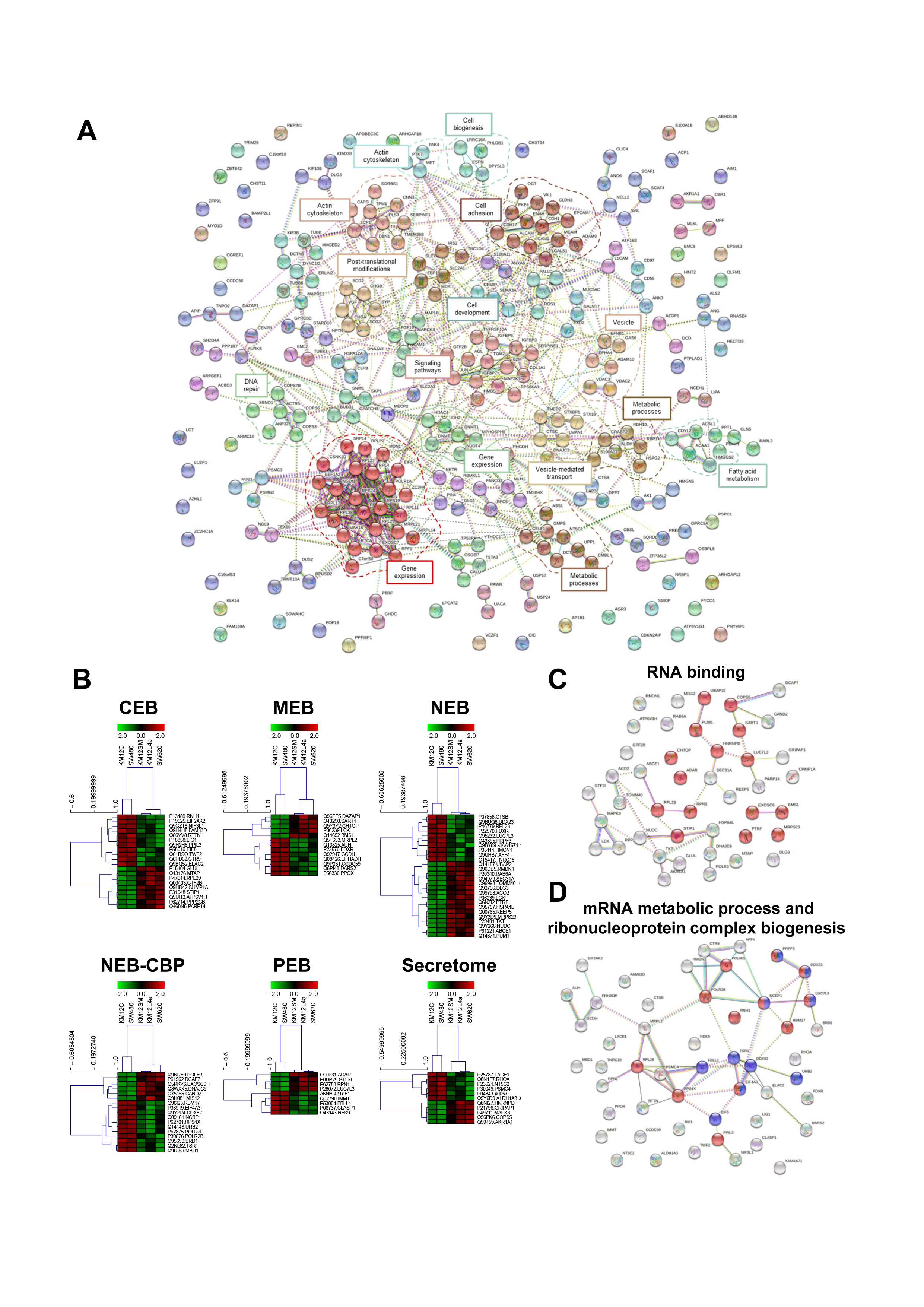
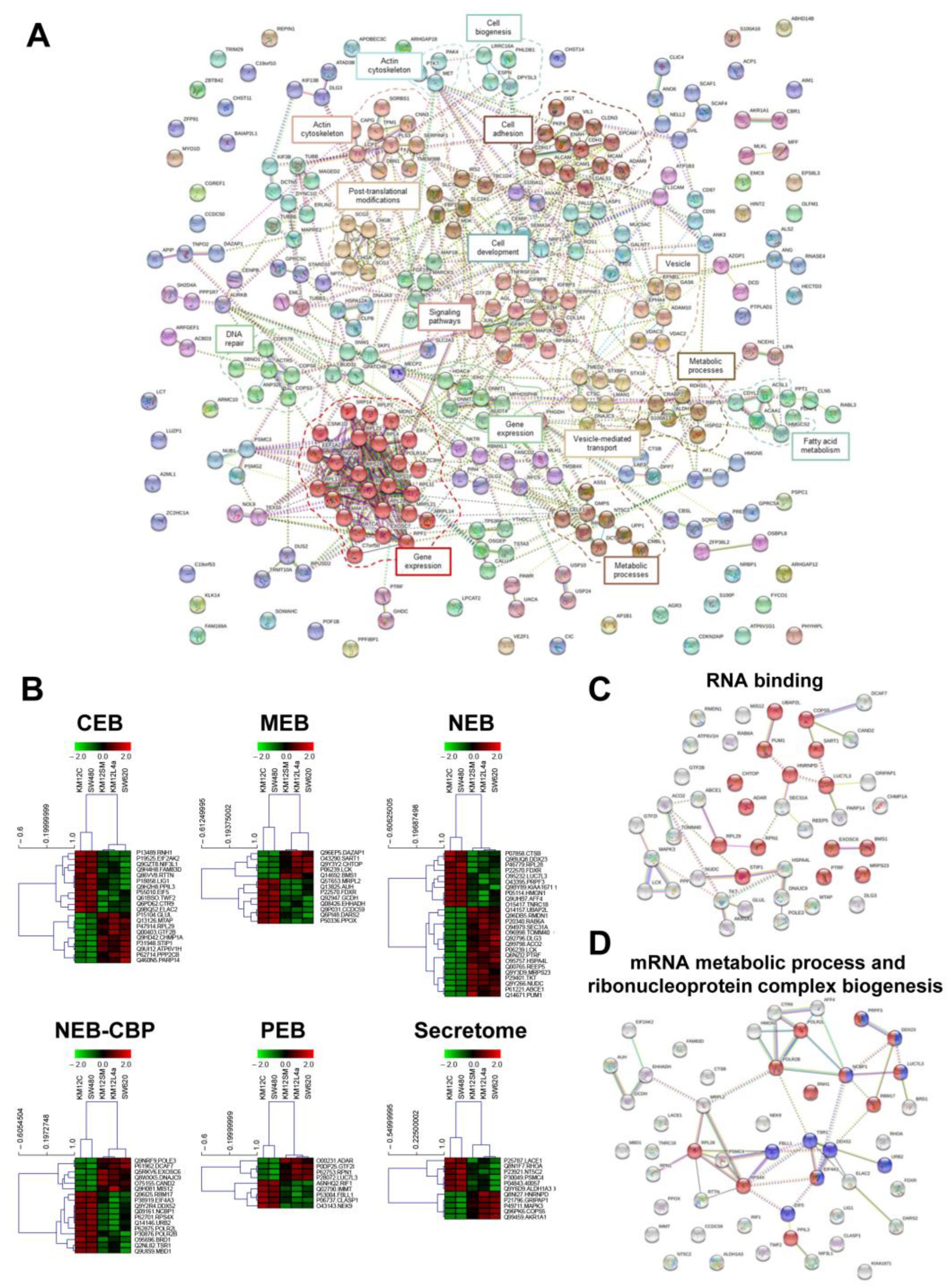
3.3. Protein clusters, Pathways and Network Analysis of Differentially Expressed Proteins in CRC Metastatic Cells
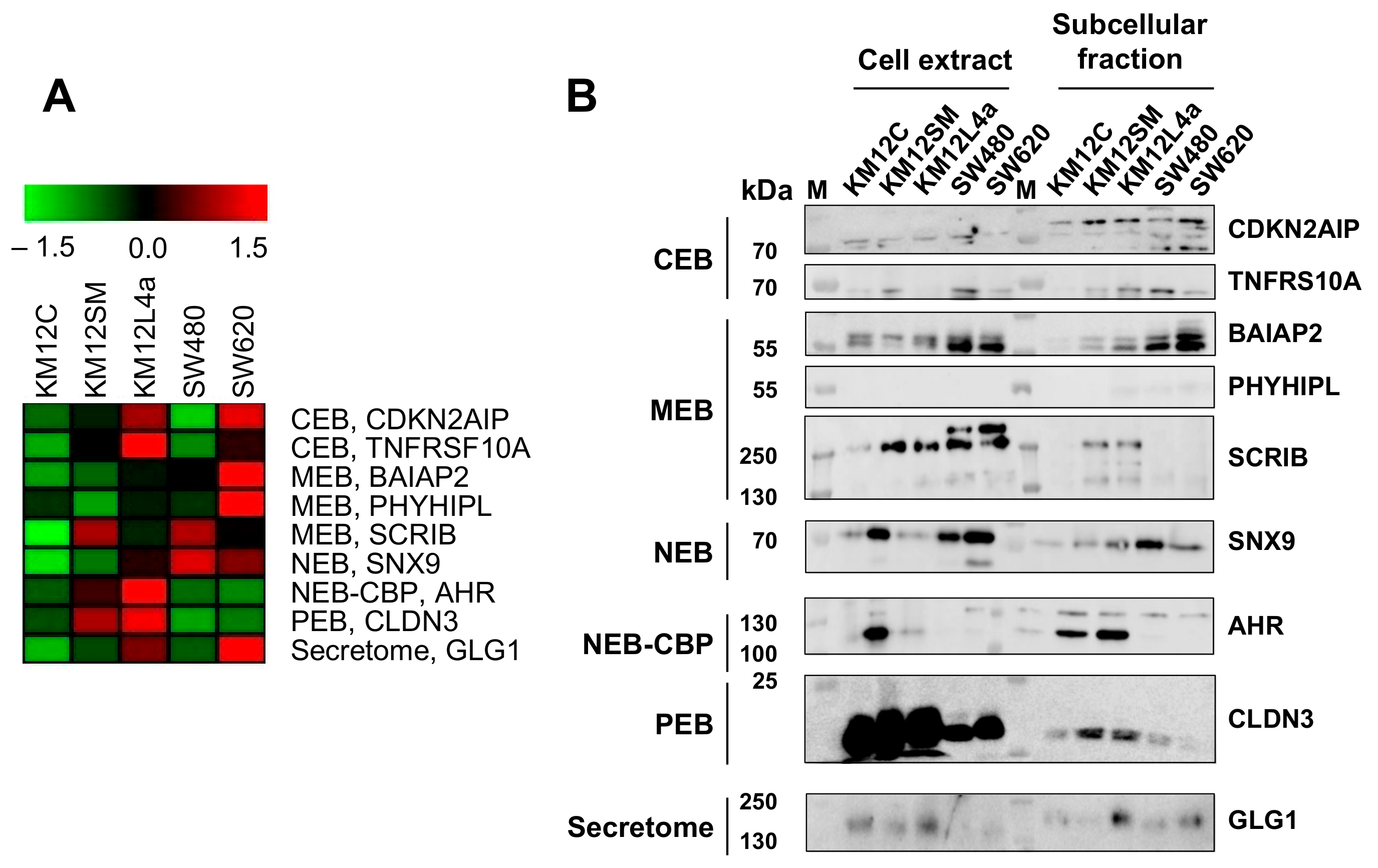
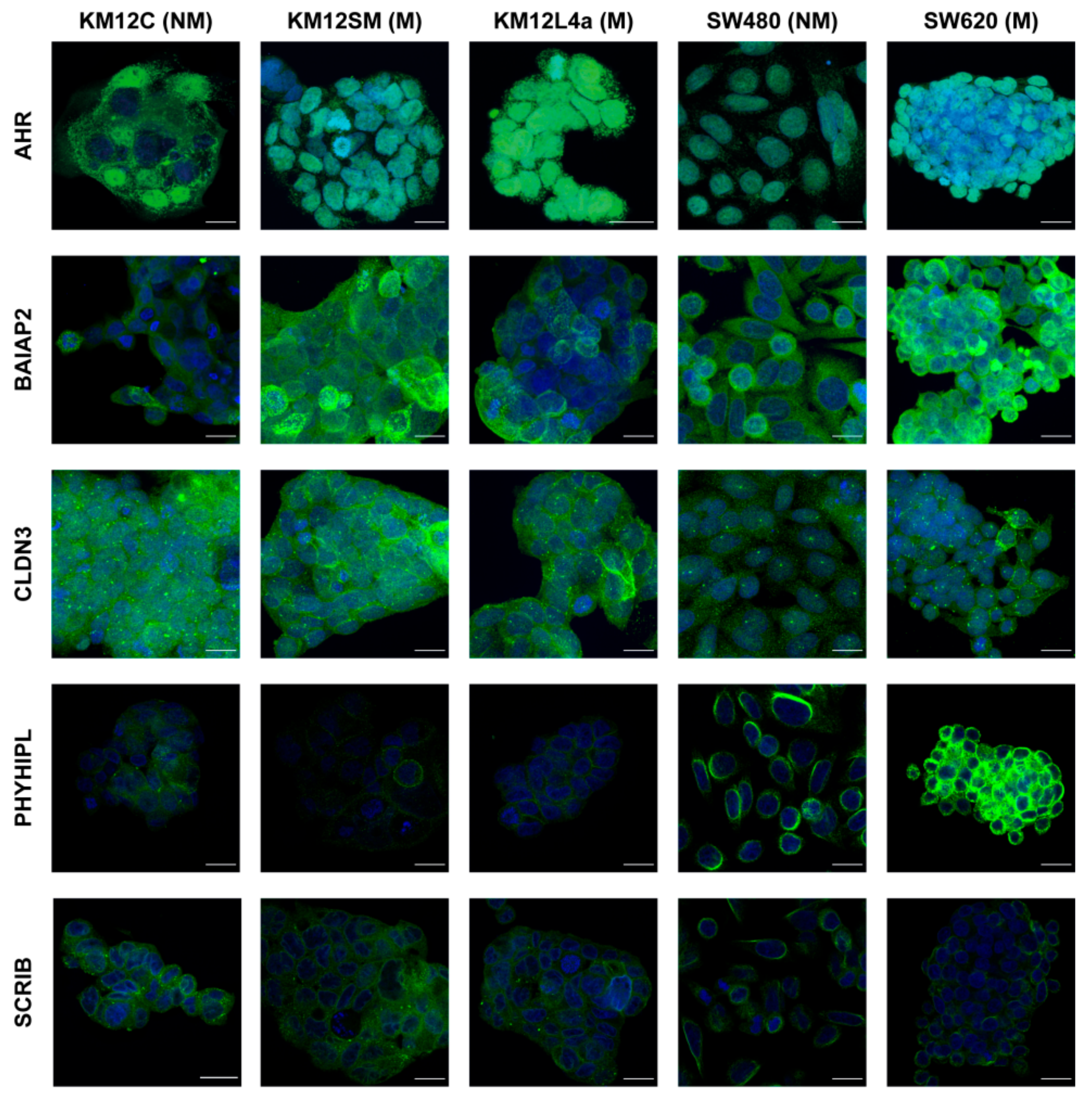
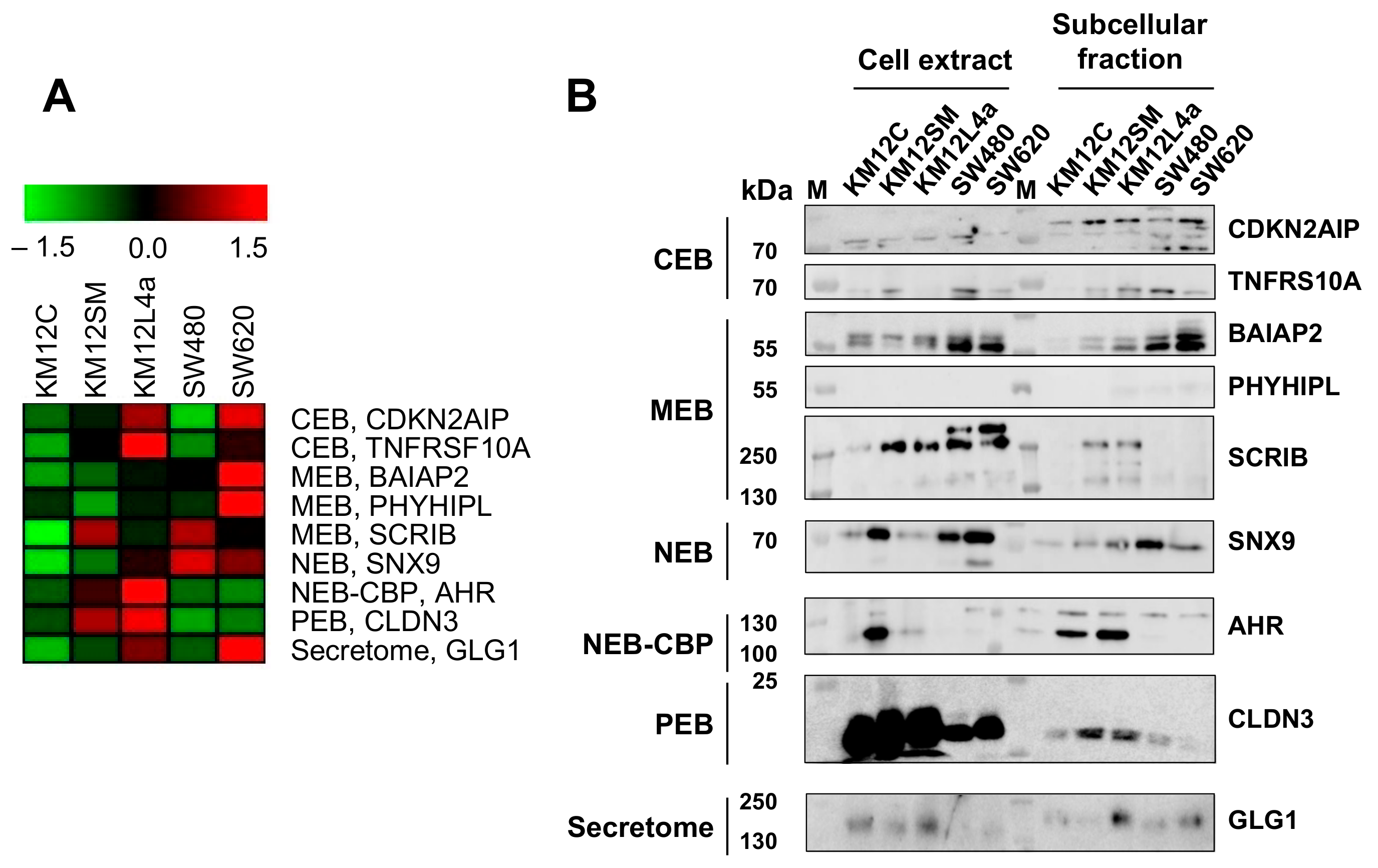
3.4. Validation of Dysregulation of Proteins in Abundance and/or Localization
3.5. Relevance of Dysregulated Proteins in Colorectal Cancer
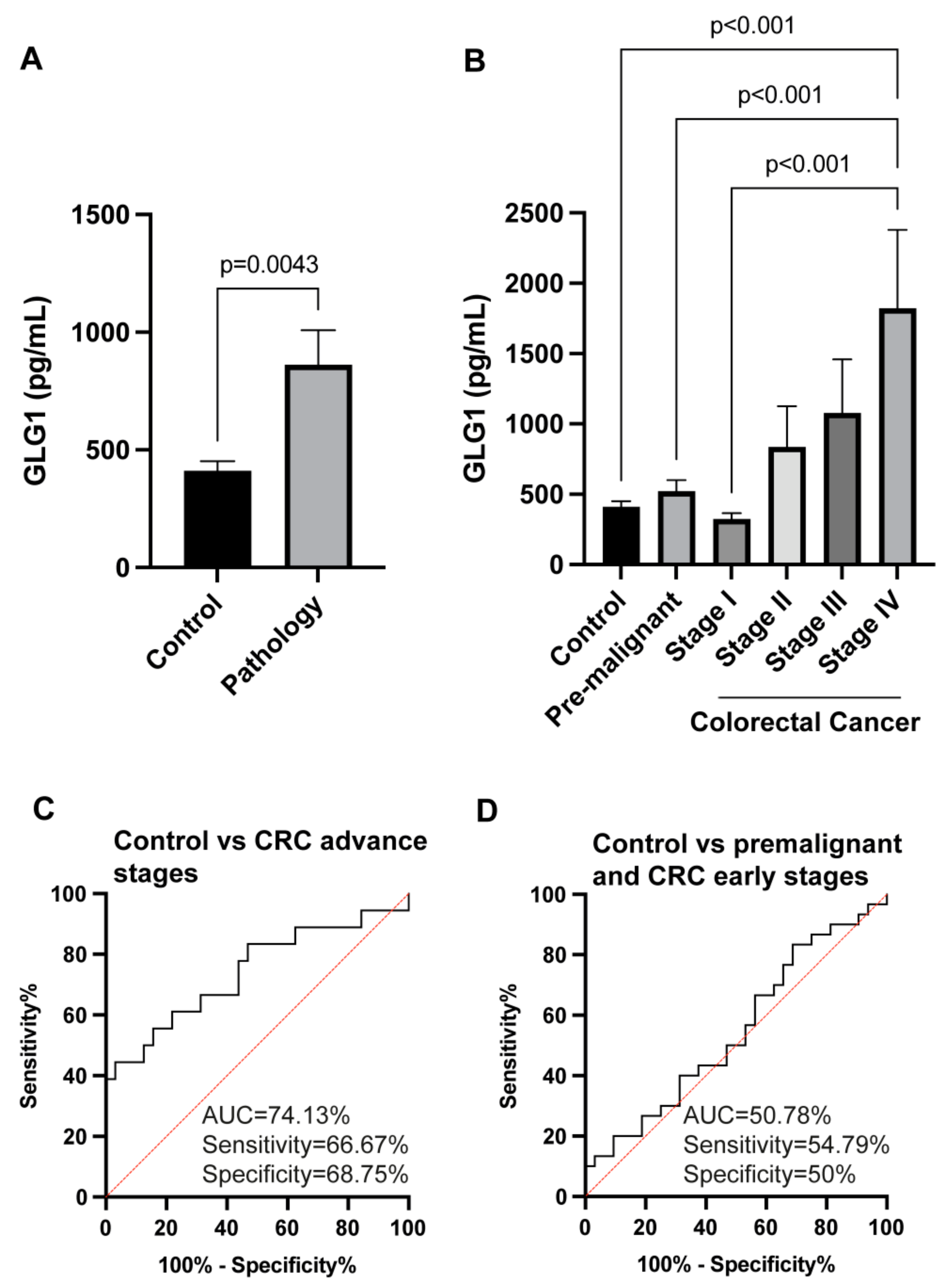
3.6. GLG1 Analysis as Blood-Based Candidate Biomarkers for Colorectal Cancer Diagnosis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rodriguez-Salas, N.; Dominguez, G.; Barderas, R.; Mendiola, M.; Garcia-Albeniz, X.; Maurel, J.; Batlle, J.F. Clinical relevance of colorectal cancer molecular subtypes. Crit. Rev. Oncol. Hematol. 2017, 109, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Holleczek, B.; Rossi, S.; Domenic, A.; Innos, K.; Minicozzi, P.; Francisci, S.; Hackl, M.; Eisemann, N.; Brenner, H. On-going improvement and persistent differences in the survival for patients with colon and rectum cancer across Europe 1999–2007–Results from the EUROCARE-5 study. Eur. J. Cancer 2015, 51, 2158–2168. [Google Scholar] [CrossRef] [PubMed]
- van der Geest, L.G.; Lam-Boer, J.; Koopman, M.; Verhoef, C.; Elferink, M.A.; de Wilt, J.H. Nationwide trends in incidence, treatment and survival of colorectal cancer patients with synchronous metastases. Clin. Exp. Metastasis 2015, 32, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Riihimaki, M.; Hemminki, A.; Sundquist, J.; Hemminki, K. Patterns of metastasis in colon and rectal cancer. Sci. Rep. 2016, 6, 29765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Hapach, L.A.; Mosier, J.A.; Wang, W.; Reinhart-King, C.A. Engineered models to parse apart the metastatic cascade. NPJ Precis. Oncol. 2019, 3, 20. [Google Scholar] [CrossRef]
- Torres, S.; Bartolome, R.A.; Mendes, M.; Barderas, R.; Fernandez-Acenero, M.J.; Pelaez-Garcia, A.; Pena, C.; Lopez-Lucendo, M.; Villar-Vazquez, R.; de Herreros, A.G.; et al. Proteome profiling of cancer-associated fibroblasts identifies novel proinflammatory signatures and prognostic markers for colorectal cancer. Clin. Cancer Res. 2013, 19, 6006–6019. [Google Scholar] [CrossRef] [Green Version]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Cai, Z.; Chiu, J.F.; He, Q.Y. Application of proteomics in the study of tumor metastasis. Genom. Proteom. Bioinform. 2004, 2, 152–166. [Google Scholar] [CrossRef] [Green Version]
- Aleckovic, M.; Wei, Y.; LeRoy, G.; Sidoli, S.; Liu, D.D.; Garcia, B.A.; Kang, Y. Identification of Nidogen 1 as a lung metastasis protein through secretome analysis. Genes Dev. 2017, 31, 1439–1455. [Google Scholar] [CrossRef] [Green Version]
- Song, P.; Bao, H.; Yu, Y.; Xue, Y.; Yun, D.; Zhang, Y.; He, Y.; Liu, Y.; Liu, Q.; Lu, H.; et al. Comprehensive profiling of metastasis-related proteins in paired hepatocellular carcinoma cells with different metastasis potentials. Proteom. Clin. Appl. 2009, 3, 841–852. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Zhang, S.; Zhang, D.; Li, Y.; Zhao, X.; Luo, Y.; Guo, Y. Identification of metastasis-related proteins and their clinical relevance to triple-negative human breast cancer. Clin. Cancer Res. 2008, 14, 7050–7059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morikawa, K.; Walker, S.M.; Jessup, J.M.; Fidler, I.J. In vivo selection of highly metastatic cells from surgical specimens of different primary human colon carcinomas implanted into nude mice. Cancer Res. 1988, 48, 1943–1948. [Google Scholar] [PubMed]
- Morikawa, K.; Walker, S.M.; Nakajima, M.; Pathak, S.; Jessup, J.M.; Fidler, I.J. Influence of organ environment on the growth, selection, and metastasis of human colon carcinoma cells in nude mice. Cancer Res. 1988, 48, 6863–6871. [Google Scholar] [PubMed]
- Li, A.; Varney, M.L.; Singh, R.K. Constitutive expression of growth regulated oncogene (gro) in human colon carcinoma cells with different metastatic potential and its role in regulating their metastatic phenotype. Clin. Exp. Metastasis 2004, 21, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Kuniyasu, H.; Ohmori, H.; Sasaki, T.; Sasahira, T.; Yoshida, K.; Kitadai, Y.; Fidler, I.J. Production of interleukin 15 by human colon cancer cells is associated with induction of mucosal hyperplasia, angiogenesis, and metastasis. Clin. Cancer Res. 2003, 9, 4802–4810. [Google Scholar] [PubMed]
- Calon, A.; Espinet, E.; Palomo-Ponce, S.; Tauriello, D.V.; Iglesias, M.; Cespedes, M.V.; Sevillano, M.; Nadal, C.; Jung, P.; Zhang, X.H.; et al. Dependency of colorectal cancer on a TGF-beta-driven program in stromal cells for metastasis initiation. Cancer Cell 2012, 22, 571–584. [Google Scholar] [CrossRef] [Green Version]
- Barderas, R.; Bartolome, R.A.; Fernandez-Acenero, M.J.; Torres, S.; Casal, J.I. High expression of IL-13 receptor alpha2 in colorectal cancer is associated with invasion, liver metastasis, and poor prognosis. Cancer Res. 2012, 72, 2780–2790. [Google Scholar] [CrossRef] [Green Version]
- Hegde, P.; Qi, R.; Gaspard, R.; Abernathy, K.; Dharap, S.; Earle-Hughes, J.; Gay, C.; Nwokekeh, N.U.; Chen, T.; Saeed, A.I.; et al. Identification of tumor markers in models of human colorectal cancer using a 19,200-element complementary DNA microarray. Cancer Res. 2001, 61, 7792–7797. [Google Scholar]
- Lundberg, E.; Borner, G.H.H. Spatial proteomics: A powerful discovery tool for cell biology. Nat. Rev. Mol. Cell Biol. 2019, 20, 285–302. [Google Scholar] [CrossRef]
- Bauer, N.C.; Doetsch, P.W.; Corbett, A.H. Mechanisms Regulating Protein Localization. Traffic 2015, 16, 1039–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Li, S. Protein mislocalization: Mechanisms, functions and clinical applications in cancer. Biochim. Biophys. Acta 2014, 1846, 13–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banworth, M.J.; Li, G. Consequences of Rab GTPase dysfunction in genetic or acquired human diseases. Small GTPases 2018, 9, 158–181. [Google Scholar] [CrossRef] [PubMed]
- Mendes, M.; Pelaez-Garcia, A.; Lopez-Lucendo, M.; Bartolome, R.A.; Calvino, E.; Barderas, R.; Casal, J.I. Mapping the Spatial Proteome of Metastatic Cells in Colorectal Cancer. Proteomics 2017, 17, 1700094. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R.; Gaugaz, F.Z. Fast and sensitive total protein and Peptide assays for proteomic analysis. Anal. Chem. 2015, 87, 4110–4116. [Google Scholar] [CrossRef] [PubMed]
- Plubell, D.L.; Wilmarth, P.A.; Zhao, Y.; Fenton, A.M.; Minnier, J.; Reddy, A.P.; Klimek, J.; Yang, X.; David, L.L.; Pamir, N. Extended Multiplexing of Tandem Mass Tags (TMT) Labeling Reveals Age and High Fat Diet Specific Proteome Changes in Mouse Epididymal Adipose Tissue. Mol. Cell Proteom. 2017, 16, 873–890. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Janeiro, A.; Ruz-Caracuel, I.; Ramon-Patino, J.L.; De Los Rios, V.; Villalba Esparza, M.; Berjon, A.; Yebenes, L.; Hernandez, A.; Masetto, I.; Kadioglu, E.; et al. Proteomic Analysis of Low-Grade, Early-Stage Endometrial Carcinoma Reveals New Dysregulated Pathways Associated with Cell Death and Cell Signaling. Cancers 2021, 13, 794. [Google Scholar] [CrossRef]
- Barderas, R.; Villar-Vazquez, R.; Fernandez-Acenero, M.J.; Babel, I.; Pelaez-Garcia, A.; Torres, S.; Casal, J.I. Sporadic colon cancer murine models demonstrate the value of autoantibody detection for preclinical cancer diagnosis. Sci. Rep. 2013, 3, 2938. [Google Scholar] [CrossRef] [Green Version]
- Babel, I.; Barderas, R.; Diaz-Uriarte, R.; Martinez-Torrecuadrada, J.L.; Sanchez-Carbayo, M.; Casal, J.I. Identification of tumor-associated autoantigens for the diagnosis of colorectal cancer in serum using high density protein microarrays. Mol. Cell Proteom. 2009, 8, 2382–2395. [Google Scholar] [CrossRef] [Green Version]
- Barderas, R.; Babel, I.; Diaz-Uriarte, R.; Moreno, V.; Suarez, A.; Bonilla, F.; Villar-Vazquez, R.; Capella, G.; Casal, J.I. An optimized predictor panel for colorectal cancer diagnosis based on the combination of tumor-associated antigens obtained from protein and phage microarrays. J. Proteom. 2012, 75, 4647–4655. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garranzo-Asensio, M.; San Segundo-Acosta, P.; Poves, C.; Fernandez-Acenero, M.J.; Martinez-Useros, J.; Montero-Calle, A.; Solis-Fernandez, G.; Sanchez-Martinez, M.; Rodriguez, N.; Ceron, M.A.; et al. Identification of tumor-associated antigens with diagnostic ability of colorectal cancer by in-depth immunomic and seroproteomic analysis. J. Proteom. 2020, 214, 103635. [Google Scholar] [CrossRef]
- Montero-Calle, A.; Aranguren-Abeigon, I.; Garranzo-Asensio, M.; Poves, C.; Fernández-Aceñero, M.J.; Martínez-Useros, J.; Sanz, R.; Dziaková, J.; Rodriguez-Cobos, J.; Solís-Fernández, G.; et al. Multiplexed biosensing diagnostic platforms detecting autoantibodies to tumor-associated antigens from exosomes released by CRC cells and tissue samples showed high diagnostic ability for colorectal cancer. Engineering 2021, 7, 1393–1412. [Google Scholar] [CrossRef]
- Garranzo-Asensio, M.; San Segundo-Acosta, P.; Martinez-Useros, J.; Montero-Calle, A.; Fernandez-Acenero, M.J.; Haggmark-Manberg, A.; Pelaez-Garcia, A.; Villalba, M.; Rabano, A.; Nilsson, P.; et al. Identification of prefrontal cortex protein alterations in Alzheimer’s disease. Oncotarget 2018, 9, 10847–10867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garranzo-Asensio, M.; Guzman-Aranguez, A.; Poves, C.; Fernandez-Acenero, M.J.; Montero-Calle, A.; Ceron, M.A.; Fernandez-Diez, S.; Rodriguez, N.; Gomez de Cedron, M.; Ramirez de Molina, A.; et al. The specific seroreactivity to Np73 isoforms shows higher diagnostic ability in colorectal cancer patients than the canonical p73 protein. Sci. Rep. 2019, 9, 13547. [Google Scholar] [CrossRef]
- Muñoz-San Martín, C.; Pérez-Ginés, V.; Torrente-Rodríguez, R.M.; Gamella, M.; Solís-Fernández, G.; Montero-Calle, A.; Pedrero, M.; Serafín, V.; Martínez-Bosch, N.; Navarro, N.; et al. Electrochemical immunosensing of Growth arrest-specific 6 in human plasma and tumor cell secretomes. Electrochem. Sci. Adv. 2021, e2100096. [Google Scholar] [CrossRef]
- Betge, J.; Schneider, N.I.; Harbaum, L.; Pollheimer, M.J.; Lindtner, R.A.; Kornprat, P.; Ebert, M.P.; Langner, C. MUC1, MUC2, MUC5AC, and MUC6 in colorectal cancer: Expression profiles and clinical significance. Virchows Arch. 2016, 469, 255–265. [Google Scholar] [CrossRef] [Green Version]
- Lakshmanan, I.; Rachagani, S.; Hauke, R.; Krishn, S.R.; Paknikar, S.; Seshacharyulu, P.; Karmakar, S.; Nimmakayala, R.K.; Kaushik, G.; Johansson, S.L.; et al. MUC5AC interactions with integrin beta4 enhances the migration of lung cancer cells through FAK signaling. Oncogene 2016, 35, 4112–4121. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.T.; He, K.C.; Pan, F.; Li, Y.; Wu, J. Prognostic value of Muc5AC in gastric cancer: A meta-analysis. World J. Gastroenterol. 2015, 21, 10453–10460. [Google Scholar] [CrossRef]
- Pothuraju, R.; Rachagani, S.; Krishn, S.R.; Chaudhary, S.; Nimmakayala, R.K.; Siddiqui, J.A.; Ganguly, K.; Lakshmanan, I.; Cox, J.L.; Mallya, K.; et al. Molecular implications of MUC5AC-CD44 axis in colorectal cancer progression and chemoresistance. Mol. Cancer 2020, 19, 37. [Google Scholar] [CrossRef] [Green Version]
- Neel, D.S.; Allegakoen, D.V.; Olivas, V.; Mayekar, M.K.; Hemmati, G.; Chatterjee, N.; Blakely, C.M.; McCoach, C.E.; Rotow, J.K.; Le, A.; et al. Differential Subcellular Localization Regulates Oncogenic Signaling by ROS1 Kinase Fusion Proteins. Cancer Res. 2019, 79, 546–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christopher, J.A.; Stadler, C.; Martin, C.E.; Morgenstern, M.; Pan, Y.; Betsinger, C.N.; Rattray, D.G.; Mahdessian, D.; Gingras, A.C.; Warscheid, B.; et al. Subcellular proteomics. Nat. Rev. Methods Primers 2021, 1, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Pelaez-Garcia, A.; Barderas, R.; Torres, S.; Hernandez-Varas, P.; Teixido, J.; Bonilla, F.; de Herreros, A.G.; Casal, J.I. FGFR4 role in epithelial-mesenchymal transition and its therapeutic value in colorectal cancer. PLoS ONE 2013, 8, e63695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, H.; Lu, B.; Zhang, J.; Wu, M.; Huang, Q.; Wu, Q.; Sheng, H.; Wu, D.; Hu, J.; Lai, M. Identification of serum biomarkers for colorectal cancer metastasis using a differential secretome approach. J. Proteome Res. 2010, 9, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Yu, H.; Tan, X.F.; Lim, T.K.; Zubaidah, R.M.; Tan, H.T.; Chung, M.C.; Lin, Q. Identification of key players for colorectal cancer metastasis by iTRAQ quantitative proteomics profiling of isogenic SW480 and SW620 cell lines. J. Proteome Res. 2011, 10, 4373–4387. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Huang, K.; Gao, C.; Lau, Q.C.; Pan, H.; Xie, K.; Li, J.; Liu, R.; Zhang, T.; Xie, N.; et al. Proteomics identification of ITGB3 as a key regulator in reactive oxygen species-induced migration and invasion of colorectal cancer cells. Mol. Cell Proteom. 2011, 10, M110.005397. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.; Huang, C.; Wu, J.; Li, J.; Hu, T.; Wang, Z.; Zhang, H.; Shao, Y.; Fu, Z. SCRIB Promotes Proliferation and Metastasis by Targeting Hippo/YAP Signalling in Colorectal Cancer. Front. Cell Dev. Biol. 2021, 9, 656359. [Google Scholar] [CrossRef]
- Tanigawa, K.; Maekawa, M.; Kiyoi, T.; Nakayama, J.; Kitazawa, R.; Kitazawa, S.; Semba, K.; Taguchi, T.; Akita, S.; Yoshida, M.; et al. SNX9 determines the surface levels of integrin beta1 in vascular endothelial cells: Implication in poor prognosis of human colorectal cancers overexpressing SNX9. J. Cell Physiol. 2019, 234, 17280–17294. [Google Scholar] [CrossRef] [Green Version]
- Cherradi, S.; Martineau, P.; Gongora, C.; Del Rio, M. Claudin gene expression profiles and clinical value in colorectal tumors classified according to their molecular subtype. Cancer Manag. Res. 2019, 11, 1337–1348. [Google Scholar] [CrossRef] [Green Version]
- Xie, G.; Raufman, J.P. Role of the Aryl Hydrocarbon Receptor in Colon Neoplasia. Cancers 2015, 7, 1436–1446. [Google Scholar] [CrossRef]
- Oda, K.; Shiratsuchi, T.; Nishimori, H.; Inazawa, J.; Yoshikawa, H.; Taketani, Y.; Nakamura, Y.; Tokino, T. Identification of BAIAP2 (BAI-associated protein 2), a novel human homologue of hamster IRSp53, whose SH3 domain interacts with the cytoplasmic domain of BAI1. Cytogenet. Cell Genet. 1999, 84, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Esposito, M.; Mondal, N.; Greco, T.M.; Wei, Y.; Spadazzi, C.; Lin, S.C.; Zheng, H.; Cheung, C.; Magnani, J.L.; Lin, S.H.; et al. Bone vascular niche E-selectin induces mesenchymal-epithelial transition and Wnt activation in cancer cells to promote bone metastasis. Nat. Cell Biol. 2019, 21, 627–639. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Ge, B.; Chen, D.; Wu, Y.; Luo, Q.; Li, X.; Zheng, C.; Tang, Q. Phytanoyl-CoA 2-Hydroxylase-Interacting Protein-Like Gene Is a Therapeutic Target Gene for Glioblastoma Multiforme. Med. Sci. Monit. 2019, 25, 2583–2590. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.S.; Kim, H.S.; Lee, S.H.; Park, W.S.; Kim, S.Y.; Park, J.Y.; Lee, J.H.; Lee, S.K.; Lee, S.N.; Jung, S.S.; et al. Mutations of tumor necrosis factor-related apoptosis-inducing ligand receptor 1 (TRAIL-R1) and receptor 2 (TRAIL-R2) genes in metastatic breast cancers. Cancer Res. 2001, 61, 4942–4946. [Google Scholar]
- Kalra, R.S.; Chaudhary, A.; Yoon, A.R.; Bhargava, P.; Omar, A.; Garg, S.; Yun, C.O.; Kaul, S.C.; Wadhwa, R. CARF enrichment promotes epithelial-mesenchymal transition via Wnt/beta-catenin signaling: Its clinical relevance and potential as a therapeutic target. Oncogenesis 2018, 7, 39. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic. Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Application | Samples (n) | Age Average ± SD (Years) | Age Range (Years) | Gender (n) | Stage | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Male | Female | Premalignant Individuals | I | II | III | IV | ||||||
| Tissue samples | WB analysis | CRC patients | 14 | 72.1 ± 12.3 | 47–88 | 5 | 9 | - | 2 | 4 | 4 | 4 |
| IHC and TMA analysis | CRC patients | 95 | 73.0 ± 9.6 | 94–51 | 57 | 38 | - | 3 | 30 | 61 | 1 | |
| Plasma samples | ELISA | CRC patients | 48 | 73.2 ± 10.3 | 49–88 | 21 | 27 | 10 | 10 | 10 | 11 | 7 |
| Healthy individuals | 32 | 57.9 ± 10.4 | 24–74 | 16 | 16 | - | ||||||
| Protein | Accession Number | Association | Compartment | Fold Change * | Up- or Downregulated |
|---|---|---|---|---|---|
| GAS6 | Q14393 | Metastatic cells | Secretome | 37.24 | Up |
| DCD | P81605 | Metastatic cells | Secretome | 35.46 | Up |
| c-MET | P08581 | Metastatic cells | Secretome | 23.31 | Up |
| LMAN1 | P49257 | Metastatic cells | Secretome | 19.17 | Up |
| CEMIP | Q8WUJ3 | Metastatic cells | Secretome | 12.39 | Up |
| HMGN5 | P82970 | Metastatic cells | PEB | 11.34 | Up |
| MUC5AC | P98088 | Metastatic cells | MEB | 9.85 | Up |
| TNFRSF10A | O00220 | Metastatic cells | CEB | 9.37 | Up |
| VIL1 | P09327 | Metastatic cells | NEB | 7.71 | Up |
| GLG1 | Q92896 | Metastatic cells | Secretome | 7.54 | Up |
| LCP1 | P13796 | Metastatic cells | CEB | 6.30 | Up |
| AGR3 | Q8TD06 | Metastatic cells | Secretome | 5.51 | Up |
| PLS3 | P13797 | Metastatic cells | Secretome | 5.33 | Up |
| CDKN2AIP | Q9NXV6 | Metastatic cells | CEB | 5.04 | Up |
| SNX9 | Q9Y5X1 | Liver and Lung metastasis | Secretome | 4.97 | Up |
| PHYHIPL | Q96FC7 | Metastatic cells | MEB | 4.07 | Up |
| ARHGAP18 | Q8N392 | Metastatic cells | NEB | 4.01 | Up |
| S100A16 | Q96FQ6 | Metastatic cells | NEB | 3.98 | Up |
| FBP1 | P09467 | Metastatic cells | CEB | 3.96 | Up |
| S100P | P25815 | Metastatic cells | Secretome | 3.30 | Up |
| PTRF | Q6NZI2 | Metastatic cells | NBP | 3.02 | Up |
| CLDN3 | O15551 | Liver and Liver and Lung metastasis | PEB | 2.57 | Up |
| AHR | P35869 | Liver and Lung metastasis | NBP | 2.52 | Up |
| SCRIB | Q14160 | Liver metastasis | MEB | 2.13 | Up |
| MAP2K3 | P46734 | Metastatic cells | NBP | 2.10 | Up |
| KIF13B | Q9NQT8 | Metastatic cells | PEB | 2.06 | Up |
| BAIAP2 | Q9UQB8 | Lymph nodes metastasis | MEB | 2.04 | Up |
| LGALS1 | P09382 | Metastatic cells | CEB | 0.20 | Down |
| PHLDB1 | Q86UU1 | Metastatic cells | MEB | 0.19 | Down |
| SLC2A1 | P11166 | Metastatic cells | MEB | 0.18 | Down |
| FYCO1 | Q9BQS8 | Metastatic cells | MEB | 0.16 | Down |
| L1CAM | P32004 | Metastatic cells | MEB | 0.16 | Down |
| ASS1 | P00966 | Metastatic cells | Secretome | 0.15 | Down |
| ICAM1 | P05362 | Metastatic cells | MEB | 0.15 | Down |
| MCAM | P43121 | Metastatic cells | MEB | 0.13 | Down |
| TGM2 | P21980 | Metastatic cells | Secretome | 0.05 | Down |
| CRABP2 | P29373 | Metastatic cells | Secretome | 0.02 | Down |
| RBP1 | P09455 | Metastatic cells | Secretome | 0.02 | Down |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Solís-Fernández, G.; Montero-Calle, A.; Martínez-Useros, J.; López-Janeiro, Á.; de los Ríos, V.; Sanz, R.; Dziakova, J.; Milagrosa, E.; Fernández-Aceñero, M.J.; Peláez-García, A.; et al. Spatial Proteomic Analysis of Isogenic Metastatic Colorectal Cancer Cells Reveals Key Dysregulated Proteins Associated with Lymph Node, Liver, and Lung Metastasis. Cells 2022, 11, 447. https://doi.org/10.3390/cells11030447
Solís-Fernández G, Montero-Calle A, Martínez-Useros J, López-Janeiro Á, de los Ríos V, Sanz R, Dziakova J, Milagrosa E, Fernández-Aceñero MJ, Peláez-García A, et al. Spatial Proteomic Analysis of Isogenic Metastatic Colorectal Cancer Cells Reveals Key Dysregulated Proteins Associated with Lymph Node, Liver, and Lung Metastasis. Cells. 2022; 11(3):447. https://doi.org/10.3390/cells11030447
Chicago/Turabian StyleSolís-Fernández, Guillermo, Ana Montero-Calle, Javier Martínez-Useros, Álvaro López-Janeiro, Vivian de los Ríos, Rodrigo Sanz, Jana Dziakova, Elena Milagrosa, María Jesús Fernández-Aceñero, Alberto Peláez-García, and et al. 2022. "Spatial Proteomic Analysis of Isogenic Metastatic Colorectal Cancer Cells Reveals Key Dysregulated Proteins Associated with Lymph Node, Liver, and Lung Metastasis" Cells 11, no. 3: 447. https://doi.org/10.3390/cells11030447
APA StyleSolís-Fernández, G., Montero-Calle, A., Martínez-Useros, J., López-Janeiro, Á., de los Ríos, V., Sanz, R., Dziakova, J., Milagrosa, E., Fernández-Aceñero, M. J., Peláez-García, A., Casal, J. I., Hofkens, J., Rocha, S., & Barderas, R. (2022). Spatial Proteomic Analysis of Isogenic Metastatic Colorectal Cancer Cells Reveals Key Dysregulated Proteins Associated with Lymph Node, Liver, and Lung Metastasis. Cells, 11(3), 447. https://doi.org/10.3390/cells11030447